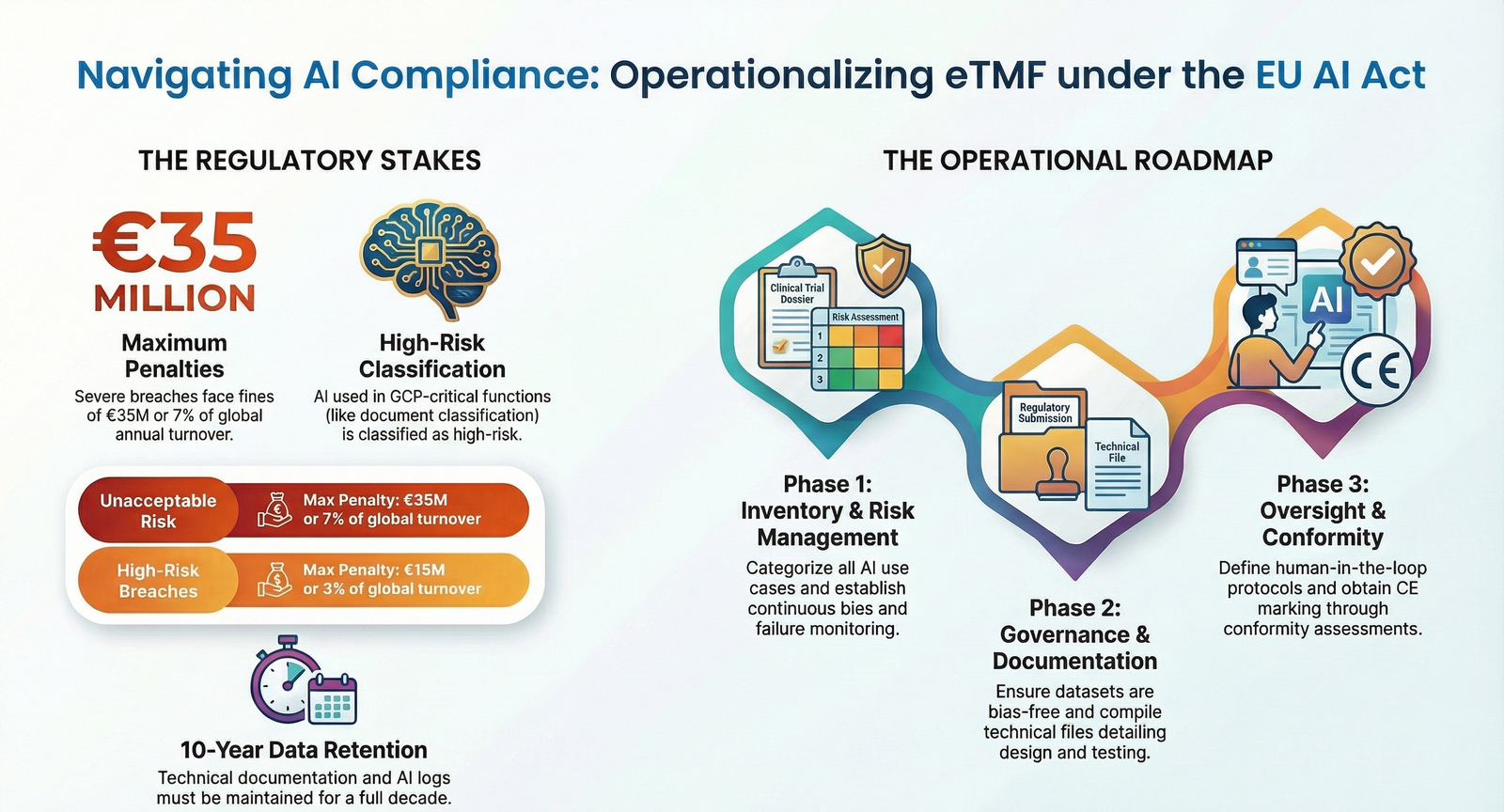
AI integration into electronic Trial Master Files (eTMF) elevates clinical research efficiency but introduces high-risk regulatory scrutiny under the EU AI Act. Operationalizing AI-enabled eTMF systems under the EU AI Act demands rigorous compliance to mitigate fines up to €35 million for unacceptable risk violations in GCP-critical clinical trial contexts.
This article outlines practical implementation steps, roles for sponsors, CROs, and vendors, and key requirements like robustness, conformity assessments, and human oversight to achieve inspection-ready AI governance.
Regulatory Landscape
Key frameworks: The EU AI Act classifies AI in GCP-critical eTMF as high-risk, mandating risk management, data governance, technical documentation, human oversight, robustness, accuracy, cybersecurity, transparency, and conformity assessments per Articles 9-23. High-risk systems require an EU Declaration of Conformity and CE marking where applicable, with integration into existing GxP, GDPR, MDR, and IVDR frameworks. Sponsors and CROs bear due diligence for procured systems.
Regulators include national market surveillance authorities, the EU AI Office, and notified bodies for assessments. Enforcement powers activate from August 2026, with penalties tiered up to €35M or 7% global turnover for unacceptable risks, €15M or 3% for high-risk breaches.
Compliance Expectations & Best Practices
Core obligations: Organizations must inventory AI components, classify risks per Annex III, maintain technical files for 10 years, implement AI quality management systems (QMS), and ensure post-market monitoring.
Vendors prepare conformity evidence; sponsors exercise oversight via quality agreements and audits. Align with ICH Q9 for risk management and 21 CFR Part 11 for data integrity.
- Conduct continuous risk assessments throughout AI lifecycle.
- Document data provenance, bias mitigation, and testing thresholds.
- Embed human review for compliance decisions.
- Integrate into broader information security frameworks.
Practical Requirements
Implementation follows a structured roadmap for sponsors, CROs, and vendors to operationalize AI-enabled eTMF compliantly.
- Step 1: Inventory and Risk Categorization – Identify all AI use cases in eTMF, classify against EU AI Act categories, focusing on GCP-critical functions like document classification or quality checks. Deliverable: Risk-classified inventory.
- Step 2: Risk Management System – Establish continuous processes per Article 9, including bias detection, failure modes, and mitigation. Integrate with QMS/ICH Q9.
- Step 3: Data Governance – Ensure datasets are representative, traceable, and bias-free per Article 10, complying with Annex 11 for audit trails.
- Step 4: Technical Documentation – Compile files detailing design, training data, testing, and logs. Append EU Declaration of Conformity (Annex V).
- Step 5: Human Oversight – Define review points, protocols, and training for AI-influenced decisions. Deliverable: SOPs and logs.
- Step 6: Transparency and Explainability – Provide user guidance on AI limits via interfaces and disclosures.
- Step 7: Conformity Assessment – Use internal or third-party (notified bodies) review; affix CE marking if required.
Common mistakes to avoid:
- Overlooking legacy AI tools in inventories.
- Inadequate bias controls in clinical data subsets.
- Skipping post-market monitoring logs.
- Failing vendor due diligence without quality agreements.
Continuous improvement: Perform regular audits, update technical files on model changes, retrain staff annually, and monitor incidents for CAPA. Leverage regulatory sandboxes for testing and EU AI Act Compliance Checkers for role-specific guidance.
Robustness, accuracy, and cybersecurity: Demonstrate consistent AI performance across studies and regions, maintain accuracy thresholds, and protect against data poisoning via GDPR-aligned security.
Why regulators intensified focus: The EU AI Act formalizes GxP-like governance for AI in clinical processes, driven by rising AI adoption in pharma and historical demands for validated systems. Enforcement pressure builds with the EU AI Office operational since 2025, targeting cross-border compliance amid digital health growth. This moment matters as high-risk obligations apply from 2027, aligning with MDR/IVDR for medical AI.
Impact on businesses: Sponsors face heightened liability for vendor AI, requiring expanded QMS, audits, and documentation. Financial exposure includes €15M fines for high-risk non-compliance; operational shifts demand AI inventories and oversight SOPs.
- Financial: Up to €35M or 7% turnover for severe breaches.
- Legal: Mandatory reporting of incidents, 10-year record retention.
- Governance: Individual accountability via training and human oversight roles.
Vendors must achieve CE marking; CROs integrate AI risks into contracts. Decision-making elevates compliance boards involving legal, IT, and data teams.
National authorities signal proactive audits on high-risk pharma AI from 2026. Industries respond with QMS extensions, notified body engagements, and voluntary GPAI codes. Experts note alignment eases burden but demands AI-specific records beyond GxP.
Pharma firms accelerate vendor qualifications; medtech adapts MDR technical files for AI Act. Market analysis predicts sandboxes aiding innovation by 2028.
As EU AI Act enforcement matures, organizations embedding these steps position for scalable compliance. Emerging standards like AI QMS and post-market tools will shape future trajectories, reducing €35M risks while advancing GCP innovation.
FAQ
1. What classifies AI in eTMF as high-risk under the EU AI Act?
Ans: AI systems integral to GCP-critical eTMF functions, such as those ensuring trial documentation compliance, qualify as high-risk per Annex III, especially if safety components of regulated products like MDR class IIa+ devices.
2. Who is responsible for conformity assessments in AI-enabled eTMF?
Ans: Providers (vendors) conduct internal or third-party assessments via notified bodies; sponsors/CROs perform due diligence and oversight to verify compliance before deployment.
3. How do fines apply to eTMF AI non-compliance?
Ans: Up to €35M or 7% global turnover for unacceptable risks; €15M or 3% for high-risk violations like inadequate risk management or documentation failures.
4. What documentation is required for high-risk eTMF AI?
Ans: Technical files covering design, data governance, testing, risk records, logs, and EU Declaration of Conformity, retained 10 years and available to authorities.
5. How to integrate human oversight in eTMF AI workflows?
Ans: Establish SOPs for review points on AI outputs affecting compliance, train personnel, and define override protocols with clinical responsibilities documented.
6. Can existing GxP frameworks satisfy EU AI Act for eTMF?
Ans: Yes, extend QMS, ICH Q9 risk processes, and technical files to cover AI-specific requirements like explainability and post-market monitoring.